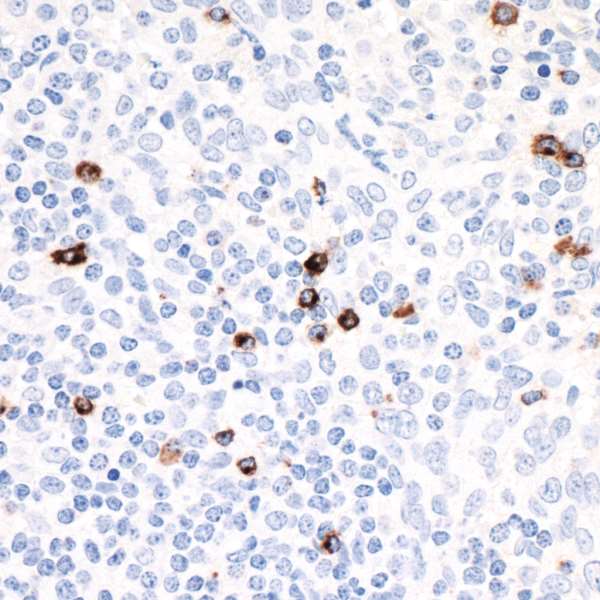
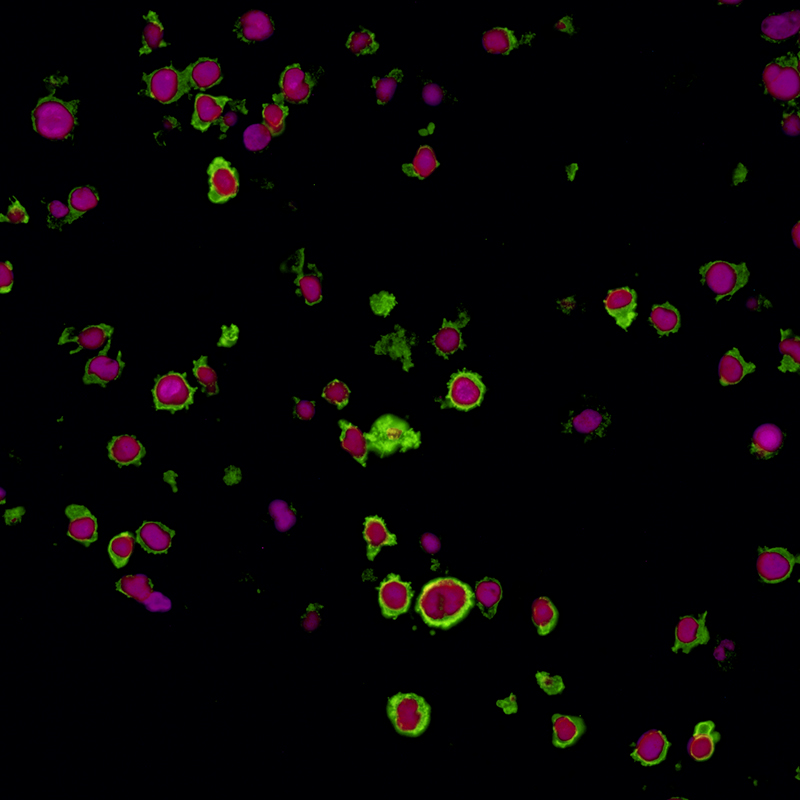
The molecular signature of exhausted T cells was first described in 2007,2 and multiple molecules involved in promotion of T cell exhaustion have since been described. Among others, these molecules include cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1), and lymphocyte activation gene 3 (LAG-3).1
LAG-3 is found on the cell surface of cytotoxic and regulatory T cells and is an immune checkpoint receptor protein that controls T cell activation and growth.3 LAG-3 is not expressed in resting T cells, but is upregulated after T cell activation in an effort to prevent autoimmunity.4 LAG-3 efficiently inhibits the immune response by binding major histocompatibility complex class II (MHC-II), which in turn inhibits T cell activation and growth.4 Expression of LAG-3 is associated with compromised antigen-specific T cell function in humans with cancer and chronic infection, but LAG-3 blocking antibodies can restore exhausted T cell function in animal models and in cultured human cells.5,6 Due to its role in regulating T cell exhaustion, LAG-3 represents a potential target for development of therapeutic agents in cancer and chronic infection.
In cancer, exhausted LAG-3-expressing cytotoxic and regulatory T cells accumulate at tumor sites,7 and in preclinical studies, LAG-3 inhibition allows for T cells to regain their cytotoxic function and subsequently decrease tumor growth.8 Clinical trials investigating LAG-3 blockade, alone or in combination with PD-1 blockade, are ongoing.9 This dual targeting of complementary immune pathways may represent a more effective strategy to reactivate previously exhausted immune response than a therapy which targets only a single receptor. In addition, clinical trials targeting a soluble form of LAG-3 which exhibits immune adjuvant activity, potentially through affecting the LAG-3/MHC-II interaction, are also underway.9 Continued development of clinical anti-LAG-3 therapies may provide a novel treatment option for patients with cancer or chronic viral infection by effectively activating (or more accurately, reactivating) the anti-tumor immune response.